Under CNDO, only the valence electrons are considered explicitly.
Interactions between electrons on the same atom are handled simply.
Since the atomic orbital basis is built from orthonormal atomic
functions, then the interaction between different orbitals ![]() and
and
![]() ,
, ![]() , due to their symmetry. The
core Hamiltonian elements,
, due to their symmetry. The
core Hamiltonian elements, ![]() , are fitted to experiment. In
CNDO/1 these were fitted to atomic ionisation energies, but in CNDO/2
this was modified by fitting to the average of the ionisation
potential and electron affinity, making the system more applicable to
both electron gain and loss.
, are fitted to experiment. In
CNDO/1 these were fitted to atomic ionisation energies, but in CNDO/2
this was modified by fitting to the average of the ionisation
potential and electron affinity, making the system more applicable to
both electron gain and loss.
In order to overcome the basis dependency of the ZDOA, the remaining
two-centre integrals only depend on their host atoms and not on the
orbital type. This is achieved by using an average electrostatic
repulsion on any atom A caused by atom B, ![]() (the `electron
repulsion integral'), so that
(the `electron
repulsion integral'), so that
![]()
In CNDO/1, ![]() is approximated by s-orbital integrals on
the atoms,
is approximated by s-orbital integrals on
the atoms,
![]()
Similarly if the core potential (consisting of the nucleus and core
electrons) on an atom B is ![]() then to maintain rotational
invariance, the interaction between an electron on atom A and this
core on atom B must be a constant, VAB (the `potential
integrals').
then to maintain rotational
invariance, the interaction between an electron on atom A and this
core on atom B must be a constant, VAB (the `potential
integrals').
Similarly to ![]() , in CNDO/1 VAB is calculated using an
s-orbital on atom A:
, in CNDO/1 VAB is calculated using an
s-orbital on atom A:
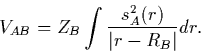
In CNDO/2, an improved parameterisation, this was replaced with a
simple approximation, ![]() .
.
The lack of monatomic differential overlap removes all terms of the form
![]()
Next the interaction between an electron and the field of two atoms,
A and B is simplified (the `resonance integral',
![]() )
)![[*]](foot_motif.gif) , by setting it proportional to the overlap integral,
, by setting it proportional to the overlap integral,
![]() ,
,
![]()
where ![]() are known as the bonding parameters and
are dependent on the atom types A and B. Under CNDO/1 these are
approximated to
are known as the bonding parameters and
are dependent on the atom types A and B. Under CNDO/1 these are
approximated to ![]() ,and
,and ![]() is fitted to ab initio calculations for each
species.
is fitted to ab initio calculations for each
species.
The precise parameterisation technique used to determine ![]() ,
,![]() , VAB,
, VAB, ![]() and
and ![]() varies.
Although I have described the method used for CNDO/1 and CNDO/2 (the
earliest parameterisations), there have been many refinements to these
since. CNDO is extremely fast, and this has meant that when computing
resources were limited, CNDO calculations have been able to perform
pioneering calculations on system sizes inaccessible to their ab
initio cousins. Much of the accuracy lost during the above
simplifications is compensated for by the use of parameterisation to
experimental and ab initio results. Although CNDO is not as
accurate as ab initio methods such as AIMPRO it remains a useful
investigative tool in semiconductor calculations, and a valuable
intermediate between empirical potentials and full ab initio
calculations.
varies.
Although I have described the method used for CNDO/1 and CNDO/2 (the
earliest parameterisations), there have been many refinements to these
since. CNDO is extremely fast, and this has meant that when computing
resources were limited, CNDO calculations have been able to perform
pioneering calculations on system sizes inaccessible to their ab
initio cousins. Much of the accuracy lost during the above
simplifications is compensated for by the use of parameterisation to
experimental and ab initio results. Although CNDO is not as
accurate as ab initio methods such as AIMPRO it remains a useful
investigative tool in semiconductor calculations, and a valuable
intermediate between empirical potentials and full ab initio
calculations.